
Quantum Information in the Protein Codes, 3-Manifolds and the Kummer Surface



1
Institut FEMTO-ST CNRS UMR 6174, Université de Bourgogne/Franche-Comté, 15 B Avenue des Montboucons, F-25044 Besançon, France
2
Quantum Gravity Research, Los Angeles, CA 90290, USA
*
Author to whom correspondence should be addressed.
Symmetry 2021, 13(7), 1146; https://doi.org/10.3390/sym13071146
Submission received: 22 April 2021
/
Revised: 6 May 2021
/
Accepted: 9 June 2021
/
Published: 26 June 2021
Abstract
:Every protein consists of a linear sequence over an alphabet of 20 letters/amino acids. The sequence unfolds in the 3-dimensional space through secondary (local foldings), tertiary (bonds) and quaternary (disjoint multiple) structures. The mere existence of the genetic code for the 20 letters of the linear chain could be predicted with the (informationally complete) irreducible characters of the finite group (with or 7 and the binary octahedral group) in our previous two papers. It turns out that some quaternary structures of protein complexes display n-fold symmetries. We propose an approach of secondary structures based on free group theory. Our results are compared to other approaches of predicting secondary structures of proteins in terms of helices, sheets and coils, or more refined techniques. It is shown that the secondary structure of proteins shows similarities to the structure of some hyperbolic 3-manifolds. The hyperbolic 3-manifold of smallest volume—Gieseking manifold—some other 3 manifolds and the oriented hypercartographic group are singled out as tentative models of such secondary structures. For the quaternary structure, there are links to the Kummer surface.
1. Introduction
We found in a previous work that the approach of quantum computation based on magic states [1,2,3] may also be used to explore the symmetries and the structure of the genetic code [4,5,6]. Given an appropriate finite group G with d conjugacy classes, one takes an irreducible character and a corresponding r-dimensional representation in the conjugacy class. For the application to the genetic code, one takes the finite group (with or 7 and the binary octahedral group) [4,5]. For such a group, the dimension r may be 1, 2, 3, 4, or 6 and the relevant conjugacy classes may be mapped to the amino acids of degeneracy r in their relation to codons. Then one defines one-dimensional projectors , where the are the states obtained from the action of a d-dimensional Pauli group on the character . When the rank of the Gram matrix with elements is , the character corresponds to a minimal informationally complete quantum measurement (or MIC), see, e.g., ([4], Section 3).
The second step of our work deals about the (secondary) genetic code found in the protein structure.
Proteins are long polymeric linear chains encoded with the 20 amino acid residues arranged in a biologically functional way. Today the protein database (or PDB) contain about entries [7]. Proteins may perform a large variety of functions in living cells and organisms including molecular recognition, catalyzing metabolic reactions, DNA replication and structural support for molecules. The sequence of amino acids leads to many different three-dimensional foldings that happen to be more conserved during evolution than the sequences themselves. The structure of proteins determines their biological function [8].
A coarse-grained representation of the backbone structure of the linear chain in a protein—a secondary code—contains three main elements that are helices and pleated sheets, due to the interactions between atoms and backbones, and random coils that indicate an absence of a regular structure. The ordered structures are held in shape by hydrogen bonds, which form between the carbonyl of one amino acid and the amino of another. In an helix, there is a pattern of bonds that puts the polypeptide chain into a helical structure with each turn of the helix containing amino acids [9]. In a pleated sheet, two or more segments of a polypeptide chain line up next to each other, forming a sheet-like structure held together by hydrogen bonds [10]. The three main elements of a protein linear chain are usually denoted H (if the segments form an helix), E (if the segments form a pleated sheet) and C (if the segments form a coil) and constitute what is called the secondary structure of the protein.
The protein secondary structure is an algebraic notation that is useful when working with X-ray diffraction and NMR structures from PDB. However in vivo proteins encounter a wide variety of effects (solvent effects, anionic and cationic concentration effects, van der Waals forces, binding to other proteins and nucleic acids) to name a few. The scheme below does lend itself to defining algebraic operations of transformations or projections that could be performed to account for some of these effects.
In this paper, we are interested in the universality of the two- or three-letter secondary code found in proteins. The letters are segments of the protein that correspond to an helix H, a pleated sheet E or a random coil C. Our view of the connection of proteins as words with two letters (or three letters) and free group theory is as follows. One defines the two-letter group or the three-letter group , where or rel(H,E,C) is the model of the protein secondary structure. For example, a hypothetical secondary code, such as , would correspond to the group which is called the modular group. Sometimes the group G corresponds (or is close in its structure) to the fundamental group of a three-dimensional manifold so that we take as a candidate manifold of the protein foldings. For the aforementioned example, the candidate manifold would be the trefoil knot complement.
We find, from several protein examples belonging to highly symmetric complexes, that the secondary code has to obey some structural algebraic constraints relying to free group theory. Our first investigation points out the possible role of two algebraic building blocks. The first one is the hyperbolic (unoriented) 3-manifold of smallest volume known as the Gieseking manifold [11], when the secondary code only consists of two letters H and C. The second one is the oriented hypercartographic group [12,13,14] (alias the two-generator free group), when the secondary code needs the three letters H, E, and C. The consistency of the (primary) genetic code and the secondary code is studied under the light of the Kummer surface that we already assumed to play a role in the quaternary structure of protein complexes [5].
In Section 2, we provide a few elements about free group theory, finitely generated subgroups of a free group and the fundamental group of a 3-manifold. We single out the mathematical objects that will be useful for our approach of the secondary structures of proteins.
In Section 3, we feature a protein example—the histone H3 of drosophila melanogaster—with a short sequence of 136 amino acids (136 aa) only comprising H and C segments in the secondary pattern. We compare the results obtained from four different models and softwares and how well they fit the cardinality sequence of subgroups of a few candidate 3-manifolds. The Gieseking manifold is a good candidate (obtained from one model) not only in terms of the cardinality sequence but also in terms of the structure of the corresponding subgroups.
In Section 4, we pass to more examples of proteins comprising H, E, and C patterns. In Section 4.1, we look at the secondary pattern of myelin P2 in homo sapiens with 133 aa. In Section 4.2, we look at the case of the gamma-carbonic anhydrase (247 aa long) within its 3-fold symmetric complex. Then, in Section 4.3, we study the Hfq protein with 74 aa in each arm of the Hfq 6-fold symmetric complex. In both cases, a theory close to the observed patterns is based on the oriented hypercartographic group , a straightforward generalization of the cartographic group introduced by A. Grothendieck in his essay [12]. In the latter case, the subgroup sequence of perfectly fits the secondary pattern of Hfq protein predicted by one particular model. In Section 4.4, we study the secondary patterns obtained for proteins belonging to 5-fold and 7-fold symmetric complexes. In particular, we provide the comparison of models for the H2A-H2B complex in nucleoplasmin and the acetylcholine receptor (with ) and the Lsm 1-7 complex (with ). In addition, one proposes a local mapping of the amino acids to a protein secondary structure with pseudo-helices, sheets and coils based on the characters of the group .
In Section 5, we investigate the nucleosome complex which is 8-fold symmetric. Following our previous work in [4,5], we find that the nucleosome complex allows to define another group theoretical model of the genetic code based on the characters of the group . In addition, one can map the DNA double helix scaffold of the nucleosome complex to the 16 singular points of a Kummer surface.
In Section 6, we briefly comment about the absolute Galois group over the rationals as an object worthwhile to be used in the context of protein sequences.
2. Algebraic Geometrical Models of Secondary Structures
Let be the free group on l generators.
It is known that every group is a quotient of some free group. One constructs a finitely presented group as the quotient of a free group G by the normal subgroup defined by a set of relations rels between the generators
One also needs to define subgroups of finite index in a group. A subgroup of the finitely presented group is generated by the words specified by a generator list that may contain words or subgroups. In the following, we are interested by the cardinality sequence that counts the number of subgroups of a finite index d up to some maximal index. This sequence allows us to identify a group (potentially as the fundamental group of a 3-manifold).
Then, to a pair corresponds the permutation group P that organizes the cosets. With the Todd-Coxeter procedure, one can obtain a permutation representation P of the pair from the action of on the coset space. In many cases, the finite group P has a geometrical meaning in the sense that it corresponds to a finite geometry [15].
Finally, the group theoretical approach may be related to the theory of 3-manifolds. According to the Poincaré conjecture (now a theorem) every simply connected closed 3-manifold is homeomorphic to the 3-sphere , alias the house of qubits [16]. However, one can dress as a 3-manifold that looses the homeomorphism to following the work of W. Thurston [17]. For instance, the three-dimensional space surrounding the tubular neighborhood of a knot—the knot complement —is a 3-manifold. Among the invariants characterizing a 3-manifold, there is the fundamental group which accounts for the first homotopy of . Finding a 3-manifold whose is the current is a way to identify the nature of the object under study.
Below we introduce two algebraic geometric objects playing a role in our description of protein secondary structures. The first object is the hyperbolic 3-manifold of the smallest volume [11,18]. The second one is the group of oriented hypermaps, a generalization of Grothendieck’s cartographic group [12,14].
2.1. The Gieseking Manifold
This 3-manifold was described by Gieseking in his 1912 thesis. One takes an ideal regular tetrahedron in the 3-dimensional hyperbolic space, that is a tetrahedron with all four vertices on the sphere at infinity and all dihedral angles equal to . Then, one identifies adjacent faces so that the orientation on the edges match ([11], Figure 1). The resulting hyperbolic manifold has minimal volume among non-compact hyperbolic manifolds. This volume is Gieseking’s constant . Remarkably, this constant also equals , which is the Dedekind zeta function at 2 for the field [18,19].
The fundamental group for the Gieseking manifold is denoted in SnapPy software [20]. The fundamental group is
The cardinality sequence of subgroups of index of is given in Table 2. The permutation groups organizing the cosets of subgroups of up to index 10 are in Table 1. The identification of sub-manifolds follows from SnapPy.
In the next section, we find that a model of the secondary structure in histone H3 (PDB 6PWE_1) (obtained with the software PORTER) is the group
It is shown in Table 1 and Table 2 that this model fits perfectly the Gieseking fundamental group at the first 7 places and approximately at the subsequent 3 places. Up to index 7 the permutation groups P are the same. At index 8, all P’s related to subgroups of are also those related to subgroups of G, but and which are related to subgroups of G are not in subgroups related to . There are also a few differences between subgroups of and G at index 9 and 10.
2.2. The Hypercartographic Group
The cartographic group is defined as
The terminology comes from Grothendieck’s Esquisse d’un programme [12,13]. It was motivated by the fact that conjugacy classes of transitive subgroups of the oriented subgroup of index 2 of the unoriented group can be identified to topological maps on connected, oriented surfaces without boundary, while more generally, conjugacy classes of can be identified with maps on connected surfaces which may or may not be orientable or have a boundary. The group was investigated by the first author in relation to quantum contextuality in quantum information [15].
Here, we are concerned with a slight generalization of the cartographic group . To interpret our results we need the oriented hypercartographic group whose definition is
This group is intimately related to the so-called Belyi’s theorem. The latter theorem states that a complex algebraic curve is defined over the field of algebraic numbers if and only if it may be uniformized by a subgroup of finite index in a triangle group. See [14] and the conclusion of the present paper for additional details.
In the section below, the group defined from the PORTER model of the secondary structure in protein Hfq (PDB 1HK9) is as follows
It is shown in Table 3 that this group perfectly fits the hypercartographic group in terms of the cardinality of subgroups up to the higher index 7 that could be calculated. In addition, the corresponding permutation groups organizing the cosets of subgroups in both the cases of and G fit as well.
2.3. Fundamental Groups of 3-Manifolds
Hyperbolic 3-manifolds that can be decomposed into regular ideal tetrahedra (up to 25 for the orientable case and up to 21 for the non-orientable case) have been investigated in [21]. Details can be found in SnapPy [20]. In Table 2 and Table 3, we collected a few 3-manifolds whose number of subgroups of index d of their fundamental group is close to that of the group arising from the secondary structure of the protein in question. For example, the figure-of-eight knot , which is the subgroup of index 2 in , corresponds to the manifold ooct_00001 in SnapPy (see Table 1 and Table 2) and is the 0-surgery on [22].
3. Secondary Structure with Helices: Drosophila Melanogaster Histone H3 (PDB 6PWE_1)
Now we show how the theory of the former section may be applied to concrete secondary structures of proteins. One starts with a simple example with two generators ( helices H and coils C). At the next section, we will study a simple example with three generators ( helices H, sheets E and coils C). Both examples are generic and provide a good credit to our models based on the unoriented hyperbolic manifold and the oriented hypercartographic group .
A review of the state of the art in the modeling of secondary structure is given in [8]. It is admitted that there is a limit imposed on the secondary structure prediction due to the somewhat arbitrary definition of three states H, E, and C. It is true that there exist other fine structures in the secondary protein pattern such as a helix, a helix and other structures belonging to DSSP (the Dictionary of Protein Secondary Structures). As a result, the assignment inconsistency would limit the highest accuracy based on three states to about 90%. In practice, the best softwares achieve a precision about 80%.
We used the softwares PSIPRED [23], PORTER 4.0 [24], PHYRE2 [25], and RAPTORX [26]. We do not enter into the details about the theory of these softwares. Below, we we find that PORTER is often well adapted to our goal of identifying an algebraic secondary structure. PORTER uses two cascaded bidirectional recurrent neural networks: one for prediction and one for filtering. The method has been trained and benchmarked by cross-validation on a set of many non redundant proteins.
3.1. The Primary (Linear) Structure
The mRNA sequence for histone H3 of drosophila melanogaster may be found in [27] with the reference NM_. It contains 529 base pairings (529 bp). A convenient way to pass from the NCBI format (with line feeds, numbers and blank spaces) to the bare linear sequence is to make use of a software such as Massager [28]. Then, a reading frame such as Expasy [29] allows to extract the candidate proteins.
The Frame 1 for sequence NM_ is as follows:
- IVFSNVK–T-TLVKPKSEMARTKQTARKSTGGKAPRKQLATKAARKSAPATGGVKKPHRYRPGTVALREIRRYQKSTELLIRKLPFQRLVREIAQDFKTDLRFQSSAVMALQEASEAYLVGLFEDTNLCAIHAKRVTIMPKDIQLARRIRGERA-ADTALTCR-SASVLYNRSFS
The partial sequence (in bold) beginning at the start codon M and ending at the stop codon ‘-’ is the histone protein H3 with the NCBI reference NP_. It can also be found at the protein data base PDB [7] with reference 6PWE_1. The sequence consists of 136 amino acids (136 aa).
3.2. The Secondary Structure
According to most models, the secondary structure of histone protein H3 only consists of subsections with an helix H or a coil C.
The predicted secondary structures obtained from the three softwares for the histone H3 protein are as follows:
- CCCCCCCCCCCCCCCCCHHHHCHHHHCCCCCCCCCCCCCCCCCCCCHHHHHHHCCCCC
- CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHCC
- CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHCCC
- CCCCCCCCCCCCCCCCCCCCHHHHHCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHCC
- HHHHHCCCCHHHHHHHHHHHCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHCHHHH
- CCHHHCCCHHHHHHHHHHHHCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHC
- HHHHHHHHHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHC
- HHHHHHHHHHHHHHHHHHCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHC
- CCCCCCHHHHHHHHHHCCCCC
- CCCCCCHHHHHHHHHHCCCCC
- CCCCCCHHHHHHHHHHCCCCC
- CCCCCCHHHHHHHHHHHCCCC
The first line is from PSIPRED, the second one is from PORTER, the third one is from PHYRE2, and the last one is from RAPTORX. One can visually check how close are the predictions.
Figure 1 is a sketch of the secondary structure of histone H3. In Table 2, it is found that the best model happens to come from the fundamental group of the Gieseking manifold described in Section 2.1.
4. Secondary Structures with Helices and Sheets: Myelin P2, Carbonic Anhydrase and the Lsm 1-7 Complex
4.1. Myelin P2 for Homo Sapiens (PDB 2WUT)
The sequence of myelin P2 in homo sapiens comprises 133 amino acids as follows. As before, the corresponding four rows for the secondary structures are from PSIPRED, PORTER, PHYRE2, and RAPTORX, respectively. One can visually check how close are the predictions.
- GMSNKFLGTWKLVSSENFDDYMKALGVGLATRKLGNLAKPTVIISKKGDIITIRTESTFKN
- CCCHHCCEEEEEEEECCHHHHHHHCCCCHHHHHHHHHCCCEEEEEEECCEEEEEEECCCC
- CCCHHCCEEEEEECCCCHHHHHHHCCCCHHHHHHHHHCCCEEEEEEECCEEEEEEECCCC
- CCCCCCEEEEEEEEECCHHHHHHHHCCCHHHHHHHHCCCCEEEEEEECCEEEEEEECCCC
- CCCCCCEEEEEEEEECCHHHHHHHCCCCHHHHHHHHCCCCEEEEEEECCEEEEEEECCCC
- TEISFKLGQEFEETTADNRKTKSIVTLQRGSLNQVQRWDGKETTIKRKLVNGKMVAECKM
- CCCHHCCEEEEEEEECCHHHHHHHCCCCHHHHHHHHHCCCEEEEEEECCEEEEEEECCCC
- EEEEEEEECCEEEEECCCCCEEEEEEEEECCEEEEEEECCCCEEEEEEEEECCEEEEEEEE
- EEEEEEECCCEEEEECCCCCEEEEEEEEECCEEEEEEECCCCCEEEEEEEECCEEEEEEEE
- EEEEEEECCCEEEEECCCCCEEEEEEEEECCEEEEEEECCCCCEEEEEEEECCEEEEEEEE
- KGVVCTRIYEKV
- CCEEEEEEEEEC
- CCEEEEEEEEEC
- CCEEEEEEEEEC
- CCEEEEEEEEEC
Figure 2 is a sketch of the secondary structure of myelin P2. Using Table 3, one observes that the cardinality sequence of subgroups in the PHYRE2 and PORTER models of the secondary structure of myelin P2 corresponds to that of the hypercartographic group up to index 4. Up to this index, one can also show that the permutation groups P for the structure of cosets in PHYRE2 and PORTER models correspond to that of .
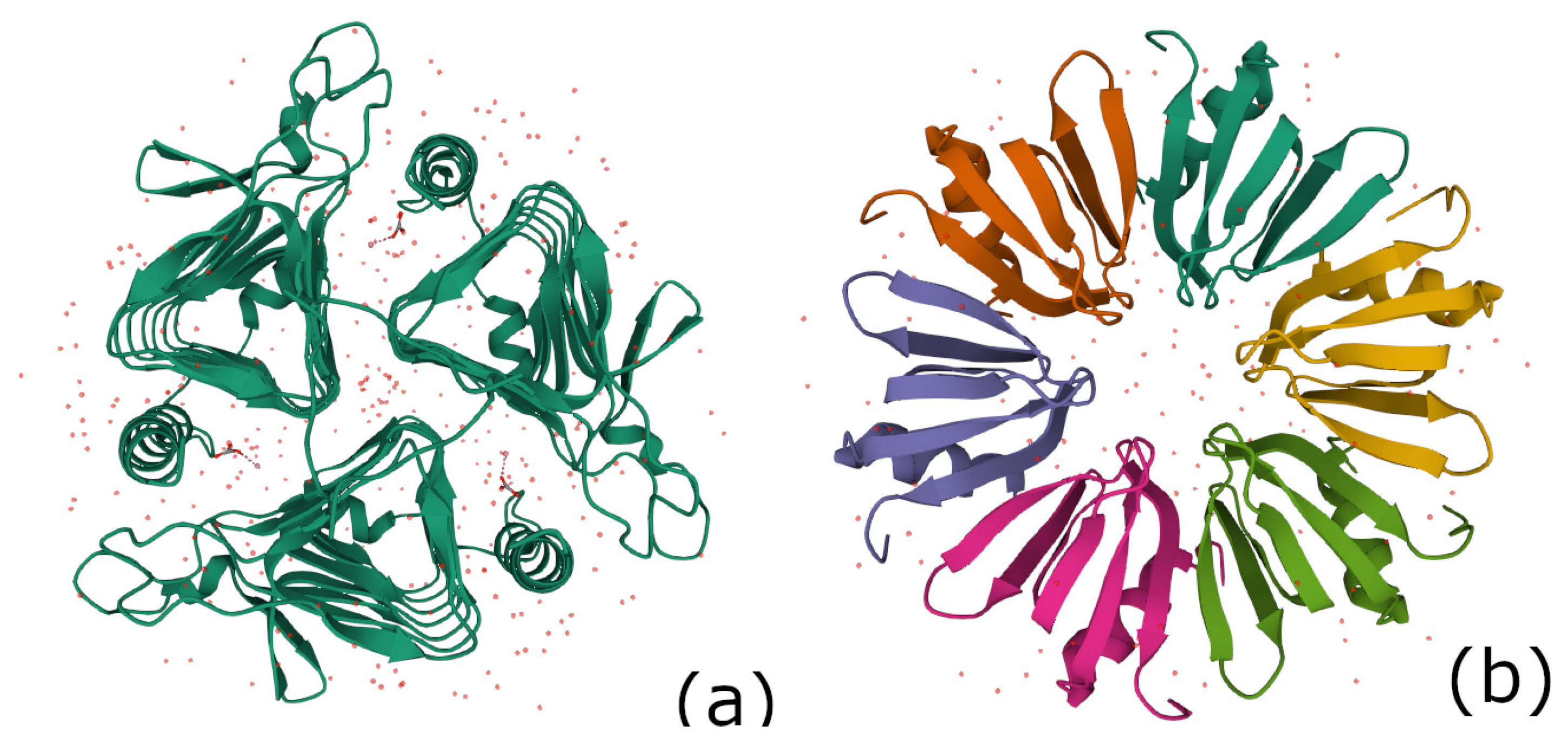
4.2. The 3-Fold Symmetric Complex for Gamma-Carbonic Anhydrase (PDB 1QRE)
In the protein data bank, the gamma-carbonic anhydrase for methanosarcina thermophila (PDB 1QRE_1) is a sequence with 247 aa. As for myelin P2, using Table 3, one observes that the cardinality sequence of subgroups in the PHYRE2 and PORTER models of the secondary structure of 1QRE_1 corresponds to that of the hypercartographic group up to index 4. The complex is 3-fold symmetric as shown in Figure 3a.
4.3. The Hfq Protein Complex of Escherichia coli (PDB 1HK9)
The sequence of Hfq protein of Escherichia coli (PDB 1HK9_1) comprises 74 amino acids. As before, the corresponding four rows for the secondary structures are from PSIPRED, PORTER, PHYRE2, and RAPTORX, respectively. One can visually check how close are the predictions.
- GAMAKGQSLQDPFLNALRRERVPVSIYLVNGIKLQGQIESFDQFVILLKNTVSQMVYKHAISTVVPSRPVSHHSCCCCCCCCCHHHHHHHHHHCCCCEEEEEECCCEEEEEEEECCCEEEEEECCCEEEEEEEEEEEEEECCCCCCCCCCCCCCCCHHHHHHHHHHHCCCCEEEEEECCEEEEEEEEEECEEEEEEECCCEEEEEEEEEEEEECCCCCCCCCCCCCCCCCCHHHHHHHHHHCCCEEEEEEECCEEEEEEEEEECCEEEEEECCCCEEEEEEEEEEEEECCEEEECCCCCCCCCCCCHHHHHHHHHCCCCEEEEECCCCEEEEEEEEECCCEEEEEECCCEEEEEEEEEEEEECCCCCCCC
The PORTER model for this protein happens to coincide with that of the hypercartographic group described in the Section 2.2.
4.4. Other n-Fold Symmetric Complexes
4.4.1. The 5-Fold Symmetric H2A-H2B Complex in Nucleoplasmin (PDB 2XQL)
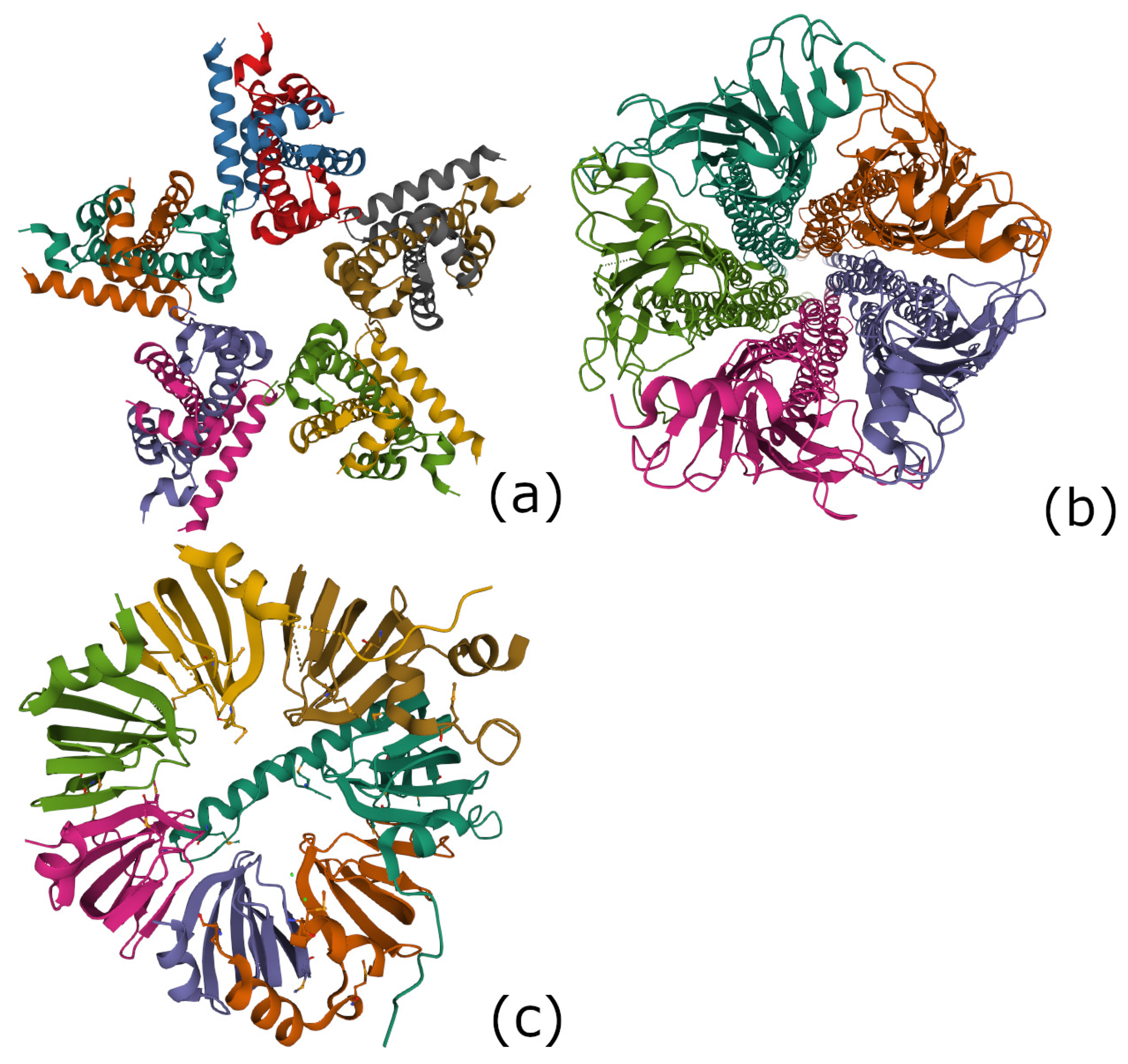
Molecular chaperones are proteins that help the folding or unfolding and the disassembly of other molecular structures. Nucleoplasmin, the first identified molecular chaperone, promotes the in vitro assembly of nucleosomes. The latter are the topic of our next section. There is a histone octamer comprising two H2A-H2B dimers and an H3-H4 tetramer. The H2A-H2B histone complex is investigated in [30]. It has a pentameric structure as shown in Figure 4a and is referred as 2XQL in the protein databank.
We performed an investigation of the secondary structure of the 2XQL_1 protein that one finds in each of the 5 arms of the complex. PSIPRED and PORTER models predict a secondary structure with helices and coils only that we could not compare to a known group theoretical sequence. The PHYRE2 and RAPTORX models, as well as our approach based on the mapping of amino acids to the characters of group and (explained below), predict a cardinality sequence which fits that of the hypercartographic group , as shown in Table 3.
4.4.2. The 5-Fold Symmetric Acetylcholine Receptor (PDB 2BG9)
The acetylcholine receptor is an integral membrane protein that responds to the binding of the acetylcholine neurotransmitter. This receptor is also sensitive to nicotine and muscarine. It has a pentameric structure shown in Figure 4b and is refereed as 2BG9 in the protein databank.
We performed an investigation of the secondary structure of the 2BG9_1 protein that one finds in the 5 arms of the complex. As shown in Table 3, all models predict a secondary structure with helices, sheets and coils. One does not observe a good fit to a group theoretical structure shared by all models. The best fit is between the RAPTORX model and the fundamental group of the 3-manifold ooct_00001 where the cardinality (and the structure) of subgroups coincide up to 4 places.
4.4.3. The 7-Fold Symmetric Lsm 1-7 Complex in the Spliceosome (PDB 4M75)
In molecular biology, there exists an ubiquitous family of RNA-binding proteins called LSM proteins whose function is to serve as scaffolds for RNA oligonucleotides, assisting the RNA to maintain the proper three-dimensional structure. Such proteins organize as rings of six or seven subunits. The Hfq protein complex was discovered in 1968 as an Escherichia coli host factor that was essential for replication of the bacteriophage [31], it displays an hexameric ring shape shown in Figure 3b of the previous subsection. As already mentioned it is remarkable that the secondary structure of Hfq protein is so close to the hypercartographic group model.
It is known that, in the process of transcription of DNA to proteins through messenger RNA sequences (mRNA), there is an important step performed in the spliceosome [32]. It includes removing the non-coding intron sequences for obtaining the exons that code for the proteinogenic amino acids. A ribonucleoprotein (RNP)—a complex of ribonucleic acid and RNA-binding protein—plays a vital role in a number of biological functions that include transcription, translation, the regulation of gene expression, and the metabolism of RNA. Individual LSm proteins assemble into a six or seven member doughnut ring which usually binds to a small RNA molecule to form a ribonucleoprotein complex.
In our previous paper [5], it was shown that 7-fold symmetry may be mirrored in the finite group (with the binary octahedral group) whose characters may be mapped to the amino acids of the genetic code. Such a mapping is reproduced in Table 4. It is important to mention that the characters of are informationally complete except for the trivial character that is not used in the mapping to amino acids and the character mapped to the starting amino acid M.
It was also determined an algebraic object called a Kummer surface playing a role in the mapping of characters to amino acids.
4.4.4. Encoding a Protein with the Characters of the Finite Group
Since the group is successful for encoding the genetic code and that, at the same time, it provides an assignment to the 20 amino acids through the corresponding characters, one can ask ourselves if may also be used to define a secondary structure in a protein. Indeed we can get a secondary structure from the character table in the following way.
Observe that, to a character in Table 4, corresponds an entry denoted , , , , , or which expresses which appears in the slot or character. This entry mainly reflects the character field associated to the character. For example, there are 11 slots (and 11 amino acids) containing and from these characters one can also define the aforementioned Kummer surface. Let us choose to assign to these slots a secondary structure and to assign a secondary structure to the remaining slots encoding an amino acid. This method allows to encode the protein under examination with pseudo-helices and pseudo-coils .
We can refine the technique by introducing more structure in the pseudo coil segments. Some of the slots/amino acids correspond to a character with constant entries and we choose to encode them as as before and the remaining slots/amino acids which correspond to a non constant entry ( or ) are encoded with , that we consider as a pseudo-sheet.
Then we can define the group
, where is the new model of the protein secondary structure obtained by our definition of pseudo-helices , pseudo-sheets , and pseudo-coils . In Table 3, the cardinality structure of group is compared to that of the other models PSIPRED, PHYRE2, PORTER, and RAPTORX. One finds that the cardinality sequence either fits, at the first few places, the hypercartographic group or that of a 3-manifold. It leaves open the question whether one of the standard models or our own model is the most efficient.
5. The 8-Fold Symmetric Histone Complex of the Nucleosome: 3WKJ in the Protein Data Bank
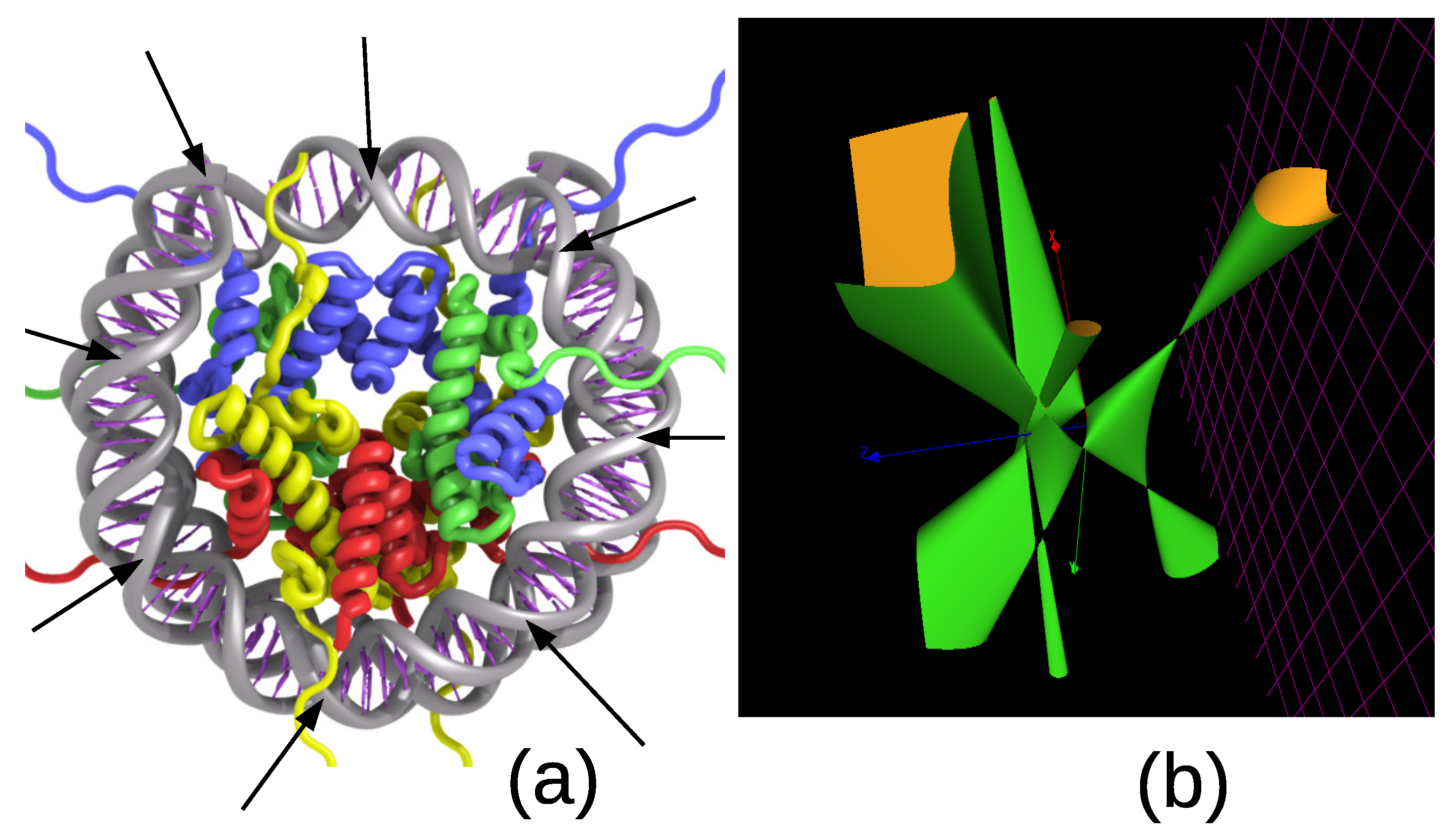
Strong DNA packaging is found in the nucleosome of eukaryotes. The nucleosome complex consists of a double helix wrapped around a set of eight histone proteins comprising two copies of H2A, H2B, H3, and H4. The nucleosome is the fundamental sub-unit of chromatin. Eukaryotic chromatin is further compacted by being folded into more complex structures eventually forming a chromosome. Nucleosomes are considered to be the support of epigenetic information. The nucleosome core particle contains approximately 146 base pairs (bp) of DNA wrapped in left-handed superhelical turns around the histone octamer as shown in Figure 5a.
We already met histone H3 of a different specie (drosophila melanogaster) in Section 3 as the preliminary example of a protein only containing helices and random coils. In the histone complex 3WKJ of the nucleosome, the secondary structure of histone H3 is also found to be made of segments with helices and coils but with a different organization according to our group theoretical approach. This is also true for the other histones H4, H2A, and H2B of the histone octamer.
In this section, we do not enter into the secondary structure of histones. We rather focus on the 8-fold symmetry of the core particle in the histone complex. What interests us about the double helix is the fact that their projection is a set of 16 double points as shown by the arrows in Figure 5a. The reader may be familiar with our previous paper [5] in which 16 double points occur in a beautiful algebraic object called a Kummer surface. Such a Kummer surface was constructed from the character table of the group in the context of the spliceosome complex that we investigated in Section 4.4. Below, we pursue in the same line of ideas and build another model of the genetic code based on the group and a corresponding Kummer surface.
The character table for the group is in Table 5. As before for the group , Table 5 contains a good assignment to the 20 amino acids and some details about the character fields through the entries . For dimensions 2 and 4, the assignments correspond to characters that are informationally complete. However, it is not the case for the assignments of amino acids in dimensions 1, 3, and 6.
All 8 characters having and in their entries are informationally complete and are at the origin of the Kummer surface. We now show an important characteristics of such characters. As an example, let us write the character number 16 as obtained from Magma [33]
where # denotes the algebraic conjugation, that is indicates replacing the root of unity w by .
One defines a genus 2 hyper-elliptic curve defined over the group from the equation
with , and . Explicitly,
leading to the polynomial definition of the Kummer surface as
The de-singularization of the Kummer surface is obtained in a simple way by restricting the product to the five first factors.
As usual for elliptic and hyper-elliptic curves of genus g, is embedded in a weighted projective plane, with weights 1, , and 1, respectively, on coordinates x, y, and z. Therefore, point triples are such that , in the field of definition, and the points at infinity take the form . Below, the software Magma is used for the calculation of points of [33]. For the points of , there is a parameter called ‘bound’ that loosely follows the heights of the x-coordinates found by the search algorithm.
It is found that the corresponding Jacobian of has points as follows:
* the 6 points bounded by the modulus 1:
, , , and .
* the 10 points of modulus :
, , , , , , , , and .
The 16 points organize as a commutative group isomorphic to the maximally abelian group as shown in the following Jacobian addition Table 6.
Where the blocks are given explicitly as
To conclude this section, we can define a model of the secondary structure of nucleosome complex based on the character table of as we did for the spliceosome complex with the character table of . The amino acids that are mapped to characters containing should belong to a pseudo-helix of the secondary structure. The other amino acids either correspond to a constant entry in the character table and belong to a pseudo-coil or to a non-constant entry (which is either , , or ) and belong to a pseudo-sheet . In Table 3, the cardinality structure of subgroups of finite index of obtained with this model is compared to that of the other models PSIPRED, PHYRE2, PORTER, and RAPTORX. One, again, observes that the cardinality sequence either fits, at the first few places, the hypercartographic group or that of a 3-manifold.
6. Discussion
The (primary) genetic code maps the 4-base words of DNA to the 20 proteinogenic amino acids, a feature that we could model by using concepts of quantum information theory associated to finite group representations. The (mostly informationally complete) characters of finite groups of signature ( the binary octahedral group) are able to account for the degeneracies and many properties of the code (see [4] when , see [5] when and Section 5 of this paper when ).
The secondary ‘genetic code’ lacks the universality of the primary code. In the standard models of the secondary structure of proteins, the mapping from the 20 amino acids to segments of helices H, sheet strands E, and coils C is not pointwise. The present generation of softwares is defined by the evolutionary information derived from alignment of multiple homologous sequences and the highest reported accuracy uses neural networks for the optimal comparison of the sequences [8].
We could identify algebraic structures in the secondary code of proteins by employing the theory of infinite groups with generators H, E, and C and the protein relation induced by the chosen model. Some hyperbolic 3-manifolds have been found as possible models of such a secondary structure. There exists a correspondence between the 3-sphere and the Bloch sphere of qubits so that a 3-manifold may be seen as a ‘dressing’ of qubits ([16], Section 1.1). In this view, quantum information controls the secondary structure. Notice that topological dynamics and negative-curvature manifolds have been proposed for modeling the brain in Reference [34].
It was unexpected that the oriented hypercartographic group seems to play a major role in the secondary structure. Why are we interested by this feature?
We are interested in geometric physical codes or languages in action [35] and their connection to the concept of emergence. Group representations arise here as a formal way to describe those geometrical codes. Back to the secondary structure of proteins, we already mentioned in the introduction that oriented hypermaps on surfaces are organized as the oriented hypercartographic group . Another important aspect is that is related to the so called absolute Galois group , the group of field-automorphisms of the field extension of the rational field . In the Esquisse d’un programme [12,13,36], Grothendieck emphasizes the interest of looking at the action of on topological, geometric and even combinatorial structures. The highest level is the so-called ‘Teichmüller tower’. The simplest level concerns bipartite (hyper)maps called ‘dessins d’enfants’. To any dessin corresponds a (so-called) Belyi function , where is a rational function of the complex variable x whose structure reflects the critical points and the topology of . The remarkable result is that acts faithfully on , that is, each non-identity element of sends two non-isomorphic dessins to two inequivalent Belyi functions , so that none of the structure of is lost by proceeding in this way. In passing, it is good to mention that the theory of ‘dessins d’enfants’ can be used to account for geometric contextuality, the counterpart of quantum contextuality [15,37].
Let us go back to the secondary structure of protein Hfq in Section 4.3 that builds one of the 7 arms of the Lsm 1-7 complex in Figure 3b. According to our theory, there is a group structure of the protein that intimately reflects that of . Every subgroup of index d of can be seen as permutation group on d elements, it can be drawn as a dessin and there is a faithful action of on all dessins and permutation groups. In other words, the protein Hfq contains in its structure the topology and algebra of . The biological meaning of this algebraic geometric structure needs further work. We leave it open at this stage. It may be that the constraint of approximating the secondary structure with three letter segments H, E, and C implies that every protein has to obey the rules. We believe that this rule may be seen as a support of the connection of biology to quantum gravity. In [38], it is shown how a theory of quantum gravity may connect to . We already proposed a connection of our approach of the genetic code (see [5] and Section 5 of this paper) to the Kummer surfaces that are surfaces and play a role in some models of quantum gravity [39].
Author Contributions
Conceptualization, M.P., F.F. and K.I.; methodology, M.P. and R.A.; software, M.P.; validation, R.A., F.F. and M.M.A.; formal analysis, M.P. and M.M.A.; investigation, M.P., F.F. and M.M.A.; writing—original draft preparation, M.P.; writing—review and editing, M.P.; visualization, F.F. and R.A.; supervision, M.P. and K.I.; project administration, K.I.; funding acquisition, K.I. All authors have read and agreed to the published version of the manuscript.
Funding
Funding was obtained from Quantum Gravity Research in Los Angeles, CA.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bartlett, S.D. Powered by magic. Nature 2014, 510, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Planat, M.; Gedik, Z. Magic informationally complete POVMs with permutations. R. Soc. Open Sci. 2017, 4, 170387. [Google Scholar] [CrossRef] [Green Version]
- Planat, M.; Aschheim, R.; Amaral, M.M.; Irwin, K. Group geometrical axioms for magic states of quantum computing. Mathematics 2019, 7, 948. [Google Scholar] [CrossRef] [Green Version]
- Planat, M.; Aschheim, R.; Amaral, M.M.; Fang, F.; Irwin, K. Complete quantum information in the DNA genetic code. Symmetry 2020, 12, 1993. [Google Scholar] [CrossRef]
- Planat, M.; Chester, D.; Aschheim, R.; Amaral, M.M.; Fang, F.; Irwin, K. Finite groups for the Kummer surface: The genetic code and quantum gravity. Quantum Rep. 2021, 3, 68–79. [Google Scholar] [CrossRef]
- Planat, M.; Aschheim, R.; Amaral, M.M.; Irwin, K. Informationally complete characters for quark and lepton mixings. Symmetry 2020, 12, 1000. [Google Scholar] [CrossRef]
- The Protein Data Bank. Available online: https://pdb101.rcsb.org/ (accessed on 1 January 2021).
- Dang, Y.; Gao, J.; Wang, J.; Heffernan, R.; Hanson, J.; Paliwal, K.; Zhou, Y. Sixty-five years of the long march in protein secondary structure prediction: The final strech? Brief. Bioinform. 2018, 19, 482–494. [Google Scholar]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauling, L.; Corey, R.B. Configurations of polypeptide chains with favored orientations around single bonds: Two new pleated sheets. Proc. Natl. Acad. Sci. USA 1951, 37, 729–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, C.C. The noncompact hyperbolic 3-manifold of minimal volume. Proc. Am. Math. Soc. 1987, 4, 100. [Google Scholar]
- Grothendieck, A. Sketch of a Programme, Written in 1984 and Reprinted with Translation in L. Schneps ans P. Lochak eds, Geometric Galois Actions 1. Around Grothendieck’s Esquisse d’un Programme, 2. The Inverse Galois Problem, Moduli Spaces and Mapping Class Groups (Cambridge University Press, 1997); (b) The Grothendieck Theory of Dessins d’Enfants, Schneps, L., Lochak, P., Eds. (Cambridge Univ. Press, 1994). Available online: https://webusers.imj-prg.fr/~leila.schneps/grothendieckcircle/EsquisseEng.pdf (accessed on 1 January 2021).
- Lando, S.K.; Zvonkin, A.K. Graphs on Surfaces and Their Applications; Springer: Berlin, Germany, 2004. [Google Scholar]
- Jones, G.; Singerman, D. Maps, hypermaps and triangle groups. In Geometric Galois Actions 1. Around Grothendieck’s Esquisse d’un Programme; Schneps, L., Lochak, P., Eds.; Cambridge University Press: Cambridge, UK, 1994; pp. 115–145. [Google Scholar]
- Planat, M.; Giorgetti, A.; Holweck, F.; Saniga, M. Quantum contextual finite geometries from dessins d’enfants. Int. J. Geom. Mod. Phys. 2015, 12, 1550067. [Google Scholar] [CrossRef] [Green Version]
- Planat, M.; Aschheim, R.; Amaral, M.M.; Irwin, K. Universal quantum computing and three-manifolds, Universal quantum computing and three-manifolds. Symmetry 2018, 10, 773. [Google Scholar] [CrossRef] [Green Version]
- Thurston, W.P. Three-Dimensional Geometry and Topology; Princeton University Press: Princeton, NJ, USA, 1997; Volume 1. [Google Scholar]
- Adams, C.C. The newest inductee in the number hall of fame. Math. Mag. 1998, 71, 341–349. [Google Scholar] [CrossRef]
- Milnor, J. Hyperbolic geometry: The first 150 years. Bull. Am. Math. Soc. 1982, 6, 9–24. [Google Scholar] [CrossRef] [Green Version]
- Culler, M.; Dunfield, N.M.; Goerner, M.; Weeks, J.R. SnapPy, a Computer Program for Studying the Geometry and Topology of 3-Manifolds. Available online: http://snappy.math.uic.edu/ (accessed on 1 January 2021).
- Fominikh, E.; Garoufalidis, S.; Goerner, M.; Tarkaev, V.; Vesnin, A. A census of tetrahedral hyperbolic manifolds. Exp. Math. 2016, 25, 466–481. [Google Scholar] [CrossRef] [Green Version]
- Planat, M.; Aschheim, R.; Amaral, M.M.; Irwin, K. Quantum computing, Seifert surfaces and singular fibers. Quantum Rep. 2019, 1, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabello, C.; Pollastri, G. Porter, PaleAle 4.0: High-accuracy prediction of protein secondary structure and relative solvent accessibility. Bioinformatics 2013, 29, 2056–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Sun, S.; Li, Z.; Zhang, R.; Xu, J. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput. Biol. 2017, 13, e1005324. [Google Scholar] [CrossRef] [Green Version]
- Genbank. Available online: https://www.ncbi.nlm.nih.gov/genbank/ (accessed on 1 January 2021).
- Nucleic Acid Sequence “Massager”. Available online: http://biomodel.uah.es/en/lab/cybertory/analysis/massager.htm (accessed on 1 January 2021).
- Translate. Available online: https://web.expasy.org/translate/ (accessed on 1 January 2021).
- Dutta, S.; Akey, I.V.; Dingwall, C.; Hartman, K.H.; Laue, T.; Nolte, R.T.; Head, J.F.; Akey, C.W. The crystal structure of nucleoplasmin-core: Implications for histone binding and nucleosome assembly. Mol. Cell 2001, 8, 841–853. [Google Scholar] [CrossRef]
- Sauter, C.; Basquin, J.; Suck, D. Sm-Like proteins in eubacteria: The crystal structure of the Hfq protein from Escherichia coli. Nucleic Acids Res. 2003, 31, 4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lührmann, W.C.L. Spliceosome, structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar]
- Bosma, W.; Cannon, J.J.; Fieker, C.; Steel, A. (Eds.) Handbook of Magma Functions, 2.23th ed.; 2017; p. 5914. Available online: http://magma.maths.usyd.edu.au/magma/ (accessed on 10 April 2021).
- Tozzi, A.; Peters, J.F.; Fingelkurts, A.A.; Marijuàn, P.C. Brain Projective Reality: Novel Clothes for the Emperor. Reply to comments on “Topodynamics of metastable brains” by Tozzi et al. Phys. Life Rev. 2017, 21, 46–55. [Google Scholar] [CrossRef]
- Irwin, K.; Amaral, M.; Chester, D. The Self-Simulation hypothesis interpretation of quantum mechanics. Entropy 2020, 22, 247. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.A. Maps on surfaces and Galois groups. Math. Slovaca 1997, 47, 1–33. [Google Scholar]
- Planat, M. Geometry of contextuality from Grothendieck’s coset space. Quantum Inf. Process. 2015, 14, 2563–2575. [Google Scholar] [CrossRef]
- Koch, R.M.; Ramgoolam, S. From matrix models and quantum fields to Hurwitz space and the absolute Galois group. arXiv 2010, arXiv:1002.1634. [Google Scholar]
- Aspinwall, P.S. K3 surfaces and string duality. In Fields, Strings and Duality, TASI 1996; Efthimiou, C., Greene, B., Eds.; World Scientific: Singapore, 1997; pp. 421–540. [Google Scholar]
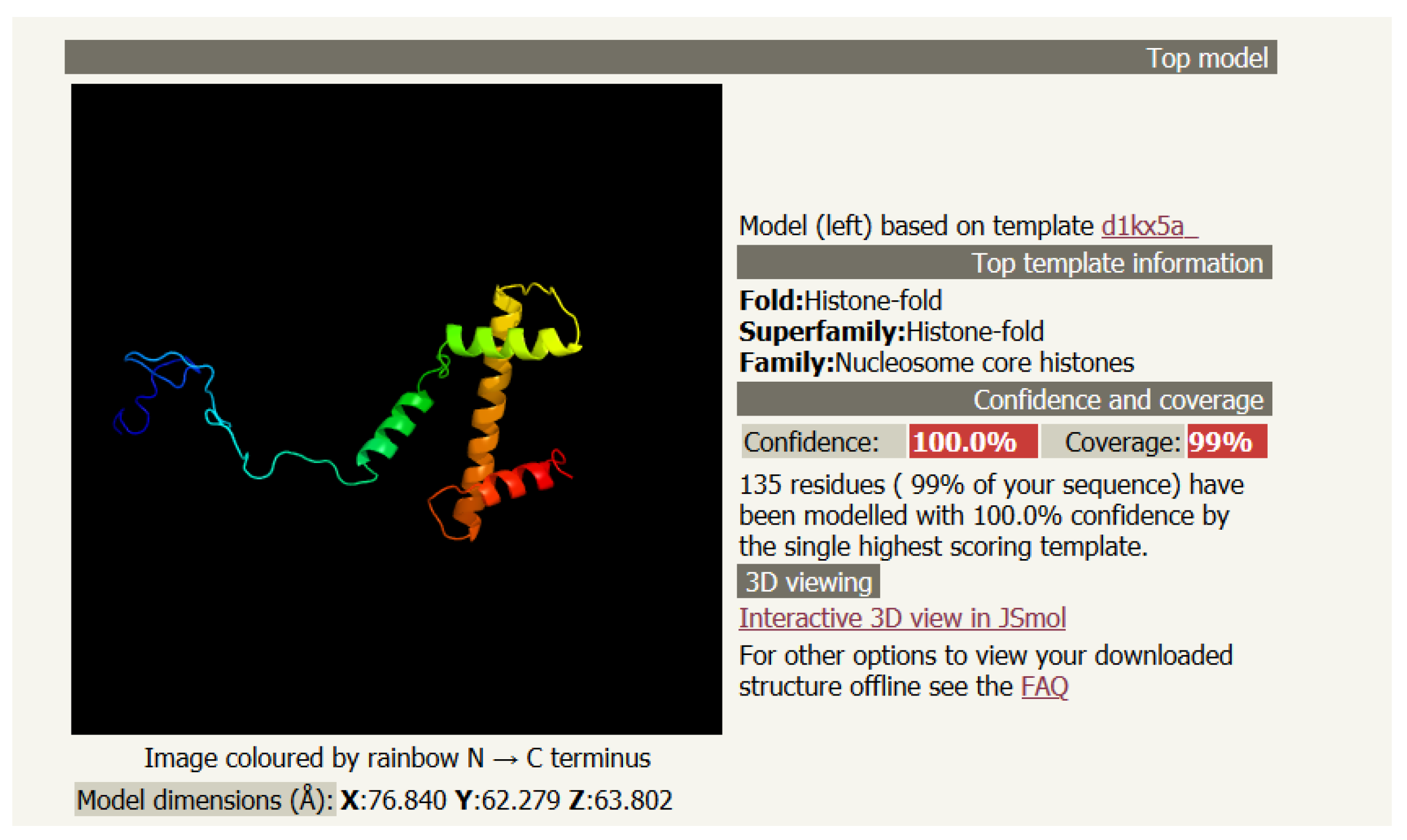
Figure 1.
A picture of the secondary structure of histone H3 as predicted from PHYRE2.

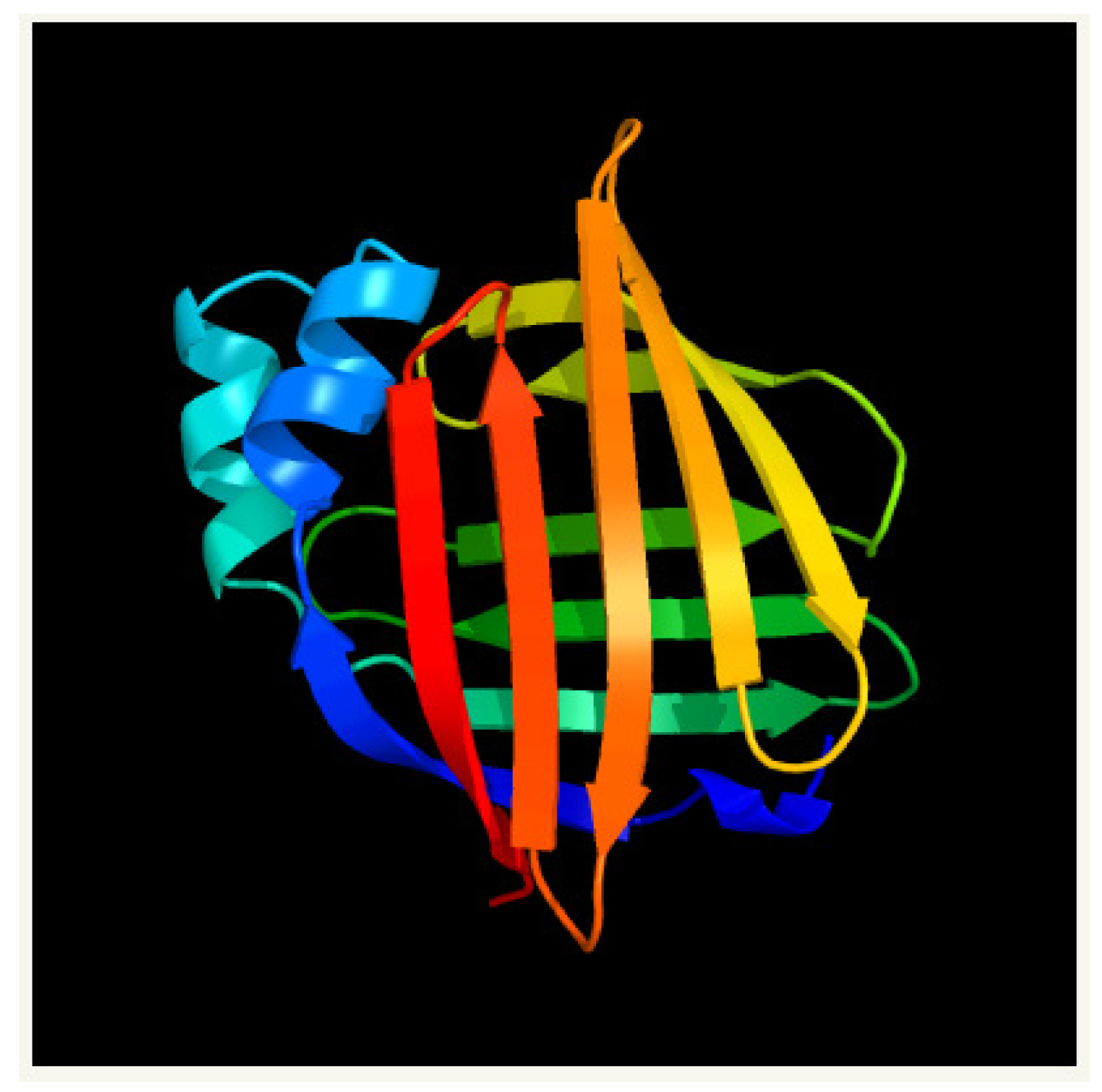
Figure 2.
A picture of the secondary structure of myelin P2 in homo sapiens (PDB 2WUT) as predicted from PHYRE2.
Figure 2.
A picture of the secondary structure of myelin P2 in homo sapiens (PDB 2WUT) as predicted from PHYRE2.

Figure 3.
(a) A picture of the structure of carbonic anhydrase (PDB 1QRE), (b) A picture of the structure of Hfq protein complex of Escherichia coli (PDB 1HK9).
Figure 3.
(a) A picture of the structure of carbonic anhydrase (PDB 1QRE), (b) A picture of the structure of Hfq protein complex of Escherichia coli (PDB 1HK9).

Figure 4.
(a) the nucleoplasmin H2A-H2B: 2XQL in the protein databank, (b) the acetylcholine receptor: 2BG9 in the protein databank, (c) the Lsm 1-7 complex in the spliceosome: 4M75 in the protein databank.
Figure 4.
(a) the nucleoplasmin H2A-H2B: 2XQL in the protein databank, (b) the acetylcholine receptor: 2BG9 in the protein databank, (c) the Lsm 1-7 complex in the spliceosome: 4M75 in the protein databank.

Figure 5.
(a) The structure of a nucleosome consists of a DNA double helix wound around eight histone proteins. There are eight periods (as shown in the picture) so that the two helices meet at 16 points. They map to the 16 double points of the Kummer surface. (b) A section at constant of the Kummer surface for the group .
Figure 5.
(a) The structure of a nucleosome consists of a DNA double helix wound around eight histone proteins. There are eight periods (as shown in the picture) so that the two helices meet at 16 points. They map to the 16 double points of the Kummer surface. (b) A section at constant of the Kummer surface for the group .


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The d-coverings () of the Gieseking manifold . The corresponding 3-manifolds (3-man) are identified thanks to SnapPy. The finite group P organizing the cosets of the index d fundamental group is given. It is shared by almost all subgroups (see lacking P) of the free group associated to the PORTER model of secondary structures of histone H3 (PDB; 6PWE_1). Some extra groups appear in the PORTER model (see extra P).
Table 1.
The d-coverings () of the Gieseking manifold . The corresponding 3-manifolds (3-man) are identified thanks to SnapPy. The finite group P organizing the cosets of the index d fundamental group is given. It is shared by almost all subgroups (see lacking P) of the free group associated to the PORTER model of secondary structures of histone H3 (PDB; 6PWE_1). Some extra groups appear in the PORTER model (see extra P).
| Index | 1 | 2 | 3 | 4 | 5 |
| 3-man | m000 | K4a1, ooct02_00001 | ntet03_00000 | m206, otet04_00002 | m407, ntet05_00007 |
| m204, ntet04_00000 | m405, ncube01_00001 | ||||
| P | (1,1) | (2,1) | (3,1) | (4,1) | (5,1) |
| (12,3) | (20,3) | ||||
| Index | 6 | 7 | 8 | 9 | 10 |
| 3-man | s961, otet06_00003 | y886, ntet07_00000 | t12839, otet06_00007 | ||
| x252, ntet06_00004 | t12840, otet08_00002 | ||||
| ntet06_00005 | ntet08_00002 | ||||
| P | (6,2) | (7,1) | (8,1) | (9,1) | (10,2) |
| (12,3) | (24,3) | ||||
| (24,13) | (24,13) | ||||
| (96,70), (192,201) | (9,1), (648,705) | (10,2), (20,3), | |||
| lacking P | (72,39) | (320,1635) | |||
| extra P | , | (216,53), , | , |
Table 2.
The models of the secondary structure for protein H3 of drosophila melanogaster and the cardinality list of d-coverings (alias conjugacy classes of subgroups) of the associated fundamental group. is the trefoil knot, is the figure-of-eight knot, the 0-surgery on is the Akbulut manifold , is the singular fiber of type II* and is the Gieseking manifold. One restricts to two-generator groups since histone H3 only consists of sections with helices and coils. Observe that the series of cardinalities for the secondary structure of H3 fits the series of the Gieseking manifolds up to the first 7 indices. Bold characters are for partial sequences matching the cardinality sequence for subgroups of the fundamental group of Gieseking manifold .
Table 2.
The models of the secondary structure for protein H3 of drosophila melanogaster and the cardinality list of d-coverings (alias conjugacy classes of subgroups) of the associated fundamental group. is the trefoil knot, is the figure-of-eight knot, the 0-surgery on is the Akbulut manifold , is the singular fiber of type II* and is the Gieseking manifold. One restricts to two-generator groups since histone H3 only consists of sections with helices and coils. Observe that the series of cardinalities for the secondary structure of H3 fits the series of the Gieseking manifolds up to the first 7 indices. Bold characters are for partial sequences matching the cardinality sequence for subgroups of the fundamental group of Gieseking manifold .
| Protein | Model | |
|---|---|---|
| H3 (6PWE_1) | PSIPRED | [1,1,1,1,2, 2,1,3,5,5 .,.,.,.,.] |
| H3 | PHYRE2 | [1,1,1,1,3, 4,1,5,10,10 .,.,.,.,.] |
| H3 | PORTER | [1,1,1,2,2, 3,1,12,6,5 .,.,.,.,.] |
| H3 | RAPTORX | [1,1,1,1,2, 1,1,2,3,3 .,.,.,.,.] |
| m000 | Gieseking | [1,1,1,2,2, 3,1,4,3,5, 4,14,1,5,10] |
| trefoil | [1,1,2,3,2, 8,7,10,18,28, 27,88,134,171,354] | |
| figure-of-eight | [1,1,1,2,4, 11,9,10,11,38, 26,62,39,89,228] | |
| (0,1) | [1,1,1,2,2, 5,1,2,2,4, 3,17,1,1,2] | |
| singular fiber II* | [1,1,2,2,1, 5,3,2,4,1, 1,12,3,3,4] |
Table 3.
A few proteins, the software used for determining their secondary structure and the cardinality list of d-coverings (alias conjugacy classes of subgroups of index d) of the associated group. One takes proteins that contain sections with helices, sheets and coils. The groups obtained by mapping the appropriate characters of and to amino acids are also considered. Bold characters are for partial sequences matching the sequence of the hypercartographic group .
Table 3.
A few proteins, the software used for determining their secondary structure and the cardinality list of d-coverings (alias conjugacy classes of subgroups of index d) of the associated group. One takes proteins that contain sections with helices, sheets and coils. The groups obtained by mapping the appropriate characters of and to amino acids are also considered. Bold characters are for partial sequences matching the sequence of the hypercartographic group .
| Protein | aa | Model | |
|---|---|---|---|
| myelin P2 (2WUT) | 133 | PSIPRED | [1, 3, 13, 84, 336, 4216] |
| 2WUT | PHYRE2 | [1, 3, 7, 26, 164, 10,669] | |
| 2WUT | PORTER | [1, 3, 7, 26, 135, 871] | |
| 2WUT | RAPTORX | [1, 3, 10, 59, 348, 2899] | |
| . | (336,118) | [1, 3, 7, 30, 122, 991] | |
| . | (384,5589) | [1, 3, 7, 34, 130, 999] | |
| carbonic anhydrase (1QRE_1) | 247 | PSIPRED | [1, 3, 10, 43, 135, 1071] |
| 1QRE_1 | PHYRE2 | [1, 3, 7, 26, 149, 1085] | |
| 1QRE_1 | PORTER | [1, 3, 7, 26, 415, 4382] | |
| 1QRE_1 | RAPTORX | [1, 3, 10, 35, 106, 804] | |
| . | (336,118) | [1, 3, 7, 30, 150, 883] | |
| . | (384,5589) | [1, 3, 10, 47, 148, 1015] | |
| protein Hfq (1HK9_1) | 74 | PSIPRED | [1, 7, 17, 114, 1145, 14,275] |
| 1HK9_1 | PHYRE2 | [1, 7, 14, 149, 1458, 21,756] | |
| 1HK9_1 | PORTER | [1, 3, 7, 26, 97, 624, 4163, 34,470] | |
| 1HK9_1 | RAPTORX | [1, 3, 10, 51, 162, 1434] | |
| . | (336,118) | [1, 3, 7, 26, 134, 912] | |
| . | (384,5589) | [1, 3, 7, 34, 146, 894] | |
| H2A-H2B (2XQL_1) | 91 | PHYRE2 | [1, 3, 7, 26, 103, 688] |
| 2XQL_1 | RAPTORX | [1, 3, 7, 26, 165, 2272] | |
| . | (336,118) | [1, 3, 7, 26, 130, 943] | |
| . | (384,5589) | [1, 3, 7, 26, 136, 967] | |
| acetylcholin receptor (2BG9_1) | 370 | PSIPRED | [1, 3, 10, 35, 151, 1023] |
| 2BG9_1 | PHYRE2 | [1, 7, 11, 92,288, 2087] | |
| 2BG9_1 | PORTER | [1, 7, 11, 92, 239, 2058] | |
| 2BG9_1 | RAPTORX | [1, 3, 7, 34, 169, 1432] | |
| . | [1, 3, 10, 47, 124, 1026] | ||
| . | [1, 3, 7, 30, 140, 931] | ||
| Lsm 1-7 complex (4M75_1) | 144 | PSIPRED | [1, 3, 16, 81, 184, 1800] |
| 4M75_1 | PHYRE2 | [1, 7, 14, 201, 705, 8850] | |
| 4M75_1 | PORTER | [1, 3, 7, 26, 139, 1118] | |
| 4M75_1 | RAPTORX | [1, 3, 7, 26, 125, 747] | |
| . | [1, 3, 7, 34, 145, 948] | ||
| . | [1, 3, 10, 35, 135, 975] | ||
| na | oriented hypermaps | [1, 3, 7, 26, 97, 624, 4163, 34,470] | |
| ooct02_00017 | 3-manifold | [1, 3, 7, 26, 40, 231] | |
| ooct02_00006 | 3-manifold | [1, 3, 10, 43, 112, 802] | |
| noct02_00024 | 3-manifold | [1, 3, 10, 43, 117, 804] | |
| ooct02_00009 | 3-manifold | [1,3,7,30,105, 649] | |
| ooct04_00001 | 3-manifold | [1, 3, 7, 34, 43, 240, 254] | |
| L7a1 | 3-manifold link | [1, 3, 7, 34, 75, 377, 807] | |
| ooct03_00019 | 3-manifold | [1, 7, 11, 85, 95, 240, 492] |
Table 4.
For the group , the table provides the dimension of the representation, the rank of the Gram matrix obtained under the action of the 29-dimensional Pauli group, the order of a group element in the class, the angles involved in the character and a good assignment to an amino acid according to its polar requirement value. All characters are informationally complete except for the trivial character and the one assigned to M. The entries involved in the characters are , , , , and featuring the angles (in ), and .
Table 4.
For the group , the table provides the dimension of the representation, the rank of the Gram matrix obtained under the action of the 29-dimensional Pauli group, the order of a group element in the class, the angles involved in the character and a good assignment to an amino acid according to its polar requirement value. All characters are informationally complete except for the trivial character and the one assigned to M. The entries involved in the characters are , , , , and featuring the angles (in ), and .
| (336,118) | dimension | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| d-dit, d = 29 | 29 | 785 | |||||||||
| amino acid | . | M | W | C | F | Y | . | . | H | Q | |
| order | 1 | 2 | 3 | 4 | 4 | 6 | 7 | 7 | 7 | 8 | |
| char | Cte | Cte | Cte | ||||||||
| polar req. | . | 5.3 | 5.2 | 4.8 | 5.0 | 5.4 | . | . | 8.4 | 8.6 | |
| (336,118) | dimension | 2 | 2 | 2 | 2 | 3 | 3 | 4 | 4 | 4 | 4 |
| d-dit, d = 29 | |||||||||||
| amino acid | N | K | E | D | I | Stop | . | . | . | . | |
| order | 14 | 14 | 14 | 21 | 21 | 21 | 21 | 21 | 21 | 21 | |
| char | Cte | Cte | Cte | ||||||||
| polar req. | 10.0 | 10.1 | 12.5 | 13.0 | 10 | 15 | . | . | . | . | |
| (336,118) | dimension | 4 | 4 | 4 | f 4 | 4 | 4 | 6 | 6 | 6 | |
| d-dit, d = 29 | |||||||||||
| amino acid | V | P | T | A | G | . | L | S | R | ||
| order | 28 | 28 | 28 | 42 | 42 | 42 | 42 | 42 | 42 | ||
| char | |||||||||||
| polar req. | 5.6 | 6.6 | 6.6 | 7.0 | 7.9 | . | 4.9 | 7.5 | 9.1 |
Table 5.
For the group , the table provides the dimension of the representation, the rank of the Gram matrix obtained under the action of the 37-dimensional Pauli group and the entries involved in the characters. The notation is , , , and . All characters having and in their entries are informationally complete and are at the origin of the Kummer surface. All characters having entries with or are also informationally complete. A good matching to the amino acids (ordered according to their polar requirement and simultaneously to the order of a group element) is given.
Table 5.
For the group , the table provides the dimension of the representation, the rank of the Gram matrix obtained under the action of the 37-dimensional Pauli group and the entries involved in the characters. The notation is , , , and . All characters having and in their entries are informationally complete and are at the origin of the Kummer surface. All characters having entries with or are also informationally complete. A good matching to the amino acids (ordered according to their polar requirement and simultaneously to the order of a group element) is given.
| (384,5589) | dimension | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 2 |
| d-dit, d = 37 | 37 | 1333 | 1333 | 1333 | 1361 | 1367 | . | ||||
| amino acid | . | . | M | W | . | . | . | . | . | . | |
| char | Cte | Cte | Cte | Cte | Cte | Cte | Cte | ||||
| (384,5589) | dimension | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 3 |
| d-dit, d = 37 | 1367 | ||||||||||
| amino acid | C | F | Y | H | Q | N | K | E | D | . | |
| char | Cte | ||||||||||
| (384,5589) | dimension | 3 | 3 | 3 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| d-dit, d = 37 | 1367 | 1367 | 1367 | 1367 | |||||||
| amino acid I | Stop | . | . | . | . | . | . | . | V | ||
| char | Cte | Cte | Cte | Cte | Cte | Cte | |||||
| (384,5589) | dimension | 4 | 4 | 4 | 4 | 6 | 6 | 6 | |||
| d-dit, d = 37 | 701 | 1365 | 1365 | ||||||||
| amino acid | P | T | A | G | L | S | R | ||||
| char | Cte |
Table 6.
The structure of the addition table for the 16 singular Jacobian points of the hyper-elliptic curves .
Table 6.
The structure of the addition table for the 16 singular Jacobian points of the hyper-elliptic curves .
| A | B | C | D |
| B | A | D | C |
| C | D | A | B |
| D | C | B | A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Planat, M.; Aschheim, R.; Amaral, M.M.; Fang, F.; Irwin, K. Quantum Information in the Protein Codes, 3-Manifolds and the Kummer Surface. Symmetry 2021, 13, 1146. https://doi.org/10.3390/sym13071146
AMA Style
Planat M, Aschheim R, Amaral MM, Fang F, Irwin K. Quantum Information in the Protein Codes, 3-Manifolds and the Kummer Surface. Symmetry. 2021; 13(7):1146. https://doi.org/10.3390/sym13071146
Chicago/Turabian StylePlanat, Michel, Raymond Aschheim, Marcelo M. Amaral, Fang Fang, and Klee Irwin. 2021. "Quantum Information in the Protein Codes, 3-Manifolds and the Kummer Surface" Symmetry 13, no. 7: 1146. https://doi.org/10.3390/sym13071146
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.